Primär biliäre Cholangitis

| Klassifikation nach ICD-10 | |
|---|---|
| K74.3 | Primäre biliäre Cholangitis Chronische nichteitrige destruktive Cholangitis |
| ICD-10 online (WHO-Version 2019) | |
Die primär biliäre Cholangitis (PBC, chronische nichteitrige destruierende Cholangitis, früher: Primär biliäre Zirrhose) ist eine relativ seltene Autoimmunerkrankung der Leber, die in ca. 90 % der Fälle Frauen betrifft. Diese beginnt (primär) an den kleinen Gallengängen (biliär), die durch eine Entzündung zerstört werden. Im längeren Verlauf kann die Entzündung auf das gesamte Lebergewebe übergreifen und zur Vernarbung führen. Da es erst im Endstadium der Erkrankung zur Zirrhose kommen kann, erfolgte in der Fachliteratur ab 2015 eine Umbenennung in „Primar biliäre Cholangitis“.[1] Die Erkrankung lässt sich häufig bereits durch antimitochondriale Antikörper (AMA) und Laborwerte wie eine erhöhte alkalische Phosphatase (AP) im Blut nachweisen, auch wenn die Leber noch relativ unversehrt ist.[2][3]
Symptome
[Bearbeiten | Quelltext bearbeiten]Besonders häufige Symptome der PBC sind Müdigkeits- und Erschöpfungszustände (70–90 % der Patienten) sowie Juckreiz (20–70 %). Rheuma-ähnliche Begleiterscheinungen umfassen Gelenkbeschwerden, Schilddrüsenerkrankungen (Hashimoto-Thyreoiditis) und trockene Schleimhäute (Sicca-Syndrom). Kleine Fetteinlagerungen in den inneren Augenwinkeln (Xanthelasmen) werden bei ca. 20 % der Patienten beobachtet. Fettstühle und Vitamin-Mangel (insbesondere Vitamin A, D, E und K) können ebenfalls mit einer PBC einhergehen. Bei 20 % der Frauen mit PBC wurden immer wieder auftretende (rezidivierende) Harnwegsinfekte gefunden. Im Spätstadium der Zirrhose können sich Zirrhose-typische Komplikationen einstellen (vgl. Zirrhose). Dazu gehören Wasserbauch (Aszites), Krampfadern in der Speiseröhre (Ösophagusvarizen) oder im Magen (Fundusvarizen), Störungen der Hirnfunktion (hepatische Enzephalopathie) und Leberkrebs. Ob die PBC auch das Risiko eines Knochenabbaus (Osteoporose) erhöht, wird durch neue Untersuchungsergebnisse jedoch in Frage gestellt.
Diagnose
[Bearbeiten | Quelltext bearbeiten]Allgemeine Laborwerte, die auf Gallenwegsentzündung bzw. Gallestau hinweisen können, sind oft erhöht: Dazu gehören die alkalische Phosphatase (AP) und die Gamma-Glutamyltranspeptidase (GGT oder Gamma-GT). Bei Leberschädigung sind auch die Leberenzyme AST und ALT erhöht, im Verlauf können die von der Leber synthetisierten Gerinnungskörper abfallen, sodass die Blutungszeit zunimmt, der Quick-Wert also abnimmt. Die Eiweißverbindung IgM (Antikörper) kann bei PBC erhöht sein.
Bei 90 % der PBC-Patienten sind die so genannten antimitochondrialen Antikörper (AMA) vom Subtyp M2 im Blut erhöht. Dieser Befund kann die Diagnose oft schon beweisen. (Eine seltene Ausnahme ohne AMA-Erhöhung ist die so genannte „AMA-negative PBC“, die auch als Autoimmuncholangitis bezeichnet wird.) Weitere PBC-spezifische antinukleäre Antikörper (ANA) richten sich gegen ‚nuclear dots‘ (sp100) und Kernmembran (gp210).
Im Ultraschall (Sonographie) kann die Leber im Frühstadium unauffällig oder ähnlich wie eine Fettleber aussehen. In späteren Stadien kann die Leber vergrößert sein, im Endstadium der Zirrhose ist die Oberfläche oft höckrig oder gewellt, die Leber kann hier wieder schrumpfen.
Eine Leberpunktion (Leberbiopsie) kann bei der Erstdiagnose helfen, die Diagnose auch über eine Gewebeuntersuchung abzusichern.
Wichtig ist es, die PBC klar von anderen Autoimmunerkrankungen wie z. B. Autoimmunhepatitis oder primär sklerosierender Cholangitis abzugrenzen. In bis zu 10 % der Fälle können auch Mischformen z. B. von PBC und Autoimmunhepatitis auftreten (so genanntes Overlap-Syndrom).
Ursachen
[Bearbeiten | Quelltext bearbeiten]PBC ist eine cholestatische Autoimmunkrankheit, deren Ursachen und Auslöser nach wie vor nicht eindeutig geklärt sind.[2][3] Bei Autoimmunkrankheiten kann das eigene Immunsystem aufgrund eines Defektes nicht mehr zwischen „Fremd“ und „Eigen“ unterscheiden und greift die Mitochondrien in körpereigenen Zellen an. Die entsprechenden Autoantikörper sind gegen die E2 Untereinheit des Pyruvat-Dehydrogenase-Komplexes, das Enzym Dihydrolipoyl-Transacetylase gerichtet.[4] Dies führt bei PBC zu einer Entzündung der kleinen Gallengänge (Cholangitis).[3]
Stark abweichende Meinungen gibt es, welche Faktoren eine PBC zum Ausbruch bringen können. Diskutiert werden u. a. hormonelle und genetische Einflüsse, Medikamente, Infektionen mit Viren, Pilzen oder Bakterien sowie Umwelteinflüsse. Eine Studie wies darauf hin, dass E2-Derivate, in denen Liponsäure durch Octin-7-carbonsäure, eine als Methylester industriell als Duft- und Geschmacksstoff (Veilchen) verwendete Verbindung, ersetzt wurde, das modifizierte E2 eine wesentlich stärkere Antigenwirkung aufwies. Allergien auf den Methylester sind bekannt. Kosmetikartikel mit diesem Stoff müssen EU-weit gekennzeichnet werden („METHYL 2-OCTYNOATE“).[5][6][7]
Der Einfluss von Schwangerschaft auf PBC und umgekehrt von PBC auf Schwangerschaft ist nicht geklärt. Alkohol ist erwiesenermaßen kein Auslöser von PBC. Er kann jedoch wie bei allen Lebererkrankungen den Verlauf ungünstig beeinflussen und sollte strikt gemieden werden.
Heutige Therapie
[Bearbeiten | Quelltext bearbeiten]Der Krankheitsverlauf ist von Patient zu Patient unterschiedlich und kann durch eine Therapie deutlich verlangsamt werden. Aktuelle Registerstudien weisen darauf hin, dass nahezu 80 % der behandelten PBC-Patienten nach 10 Jahren weiterhin am Leben sind. Historische Studien mit unbehandelten Patienten zeigten dagegen ein erhöhtes Sterberisiko von durchschnittlich 10 bis 16 Jahren nach der Erstdiagnose.[3] In Frühstadien der Erkrankung scheint die Lebenserwartung nicht wesentlich eingeschränkt zu sein. Die Standardtherapie der PBC besteht heute in Ursodeoxycholsäure (UDCA), die als Tablette gegeben werden kann. Die Therapie wird gewöhnlich gut vertragen, setzt nach Diagnosestellung ein und dauert lebenslang. Ziel der Therapie ist, den Verlauf der Erkrankung zu verlangsamen und Laborwerte zu verbessern.
Als weitere Therapie war seit 2016 die Obeticholsäure (OCA) zugelassen: entweder in Kombination mit UDCA, falls UDCA allein nicht ausreichend wirksam war, oder als Monotherapie, falls UDCA nicht toleriert wurde.[3] Im Juni 2024 hat die Europäische Arzneimittel-Agentur (EMA) jedoch empfohlen, die Zulassung von Obeticholsäure zurückzunehmen, da deren klinischer Nutzen nicht bestätigt werden konnte. Grund für diese Entscheidung der EMA sind die Ergebnisse der klinischen Studie 747-302 (COBALT)[8] an Patienten mit PBC. Aus diesen Studienergebnissen ergibt sich gemäß EMA ein negatives Nutzen-Risiko-Verhältnis von Obeticholsäure. Laut EMA sollen daher außerhalb von klinischen Studien keine neuen Patienten mit Obeticholsäure behandelt werden; für Patienten, die bereits mit Obeticholsäure behandelt werden, sollten alternative Behandlungsoptionen in Betracht gezogen werden.[9][10]
Transplantation
[Bearbeiten | Quelltext bearbeiten]Schreitet die Erkrankung trotz Therapie bis zur dekompensierten Zirrhose voran oder werden Symptome wie z. B. Juckreiz trotz Behandlung für Patienten unerträglich, wird oft eine Lebertransplantation nötig. Bei 75 % der transplantierten Patienten ist damit auch die PBC geheilt, bei 25 % können sich in der neuen Leber wieder PBC-ähnliche Schäden entwickeln. Nach einer erfolgreichen Lebertransplantation ist die Langzeitprognose von PBC-Patienten gut.
Zukunftsaussichten
[Bearbeiten | Quelltext bearbeiten]Ein Durchbruch bei der Ursachensuche der PBC ist auch in den kommenden Jahren kaum zu erwarten. Viele Studien scheinen für sich genommen plausibel, widersprechen sich aber in den Ergebnissen. Auch Meldungen in der medizinischen Fachpresse, die Ursache sei gefunden (z. B. Umwelteinflüsse oder Retroviren), sollten daher mit großer Vorsicht aufgenommen werden.
Wenn die Standardtherapie mit Ursodeoxycholsäure (UDCA) nicht ausreichend anspricht, wird untersucht, ob die Wirksamkeit durch die Kombination mit weiteren Substanzen verbessert werden kann. Zu diesem Zweck ist seit 2016 die Kombination aus UDCA und Obeticholsäure als Second-Line-Therapie zugelassen. Weitere Kombinationstherapien wie z. B. UDCA und Fibrate werden mitunter als „off-label“-Therapien eingesetzt, sind jedoch aktuell nicht zur PBC-Behandlung zugelassen (Stand: Januar 2021). In Studien werden mittlerweile auch weitere neuartige Substanzen als Kombinationspartner mit UDCA untersucht.[3][11][2]
Bildergalerie
[Bearbeiten | Quelltext bearbeiten]-
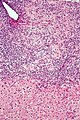 Mikrofoto einer primär biliären Zirrhose (HE-Färbung, mittlere Vergrößerung).
Mikrofoto einer primär biliären Zirrhose (HE-Färbung, mittlere Vergrößerung). -
 Mikrofoto einer primär biliären Zirrhose (HE-Färbung, niedrige Vergrößerung).
Mikrofoto einer primär biliären Zirrhose (HE-Färbung, niedrige Vergrößerung).


Siehe auch
[Bearbeiten | Quelltext bearbeiten]Literatur
[Bearbeiten | Quelltext bearbeiten]- U. Leuschner: Autoimmunkrankheiten der Leber und Overlapsyndrome. 1. Auflage. UNI-MED Verlag, 2001.
- H. Rautiainen u. a.: Budesonide Combined with UDCA to Improve Liver Histology in Primary Biliary Cirrhosis: A Three-Year Randomized Trial. In: Hepatology, April 2005, S. 747–752.
- A. S. Abdulkarim, L. M. Petrovic, W. R. Kim, P. Angulo, R. V. Lloyd, K. D. Lindor: Primary biliary cirrhosis: an infectious disease caused by Chlamydia pneumoniae? In: Journal of Hepatology, März 2004, 40(3), S. 380–384.
- K. Dohmen u. a.: Atrophic corpus gastritis and Helicobacter pylori infection in primary biliary cirrhosis. In: Dig Dis Sci. Januar 2002, 47(1), S. 162–169.
- M. Durazzo u. a.: Lack of association between seroprevalence of Helicobacter pylori infection and primary biliary cirrhosis. In: World J Gastroenterol., 1. November 2004, 10(21), S. 3179–3181.
- A. Floreani, A. Mega, V. Camozzi, V. Baldo, M. Plebani, P. Burra, G. Luisetto: Is osteoporosis a peculiar association with primary biliary cirrhosis? In: World J Gastroenterol., 14. September 2005, 11(34), S. 5347–5350.
- M. Gershwin u. a.: Apocalypsal versus Apocryphal: The Role of Retroviruses in Primary Biliary Cirrhosis. In: American Journal of Gastroenterology, Dezember 2004, Volume 99, Issue 12, S. 2356.
- R. Klein, M. Wiebel, S. Engelhart, P. A. Berg: Sera from patients with tuberculosis recognize the M2a-epitope (E2-subunit of pyruvate dehydrogenase) specific for primary biliary cirrhosis. In: Clin Exp Immunol., Mai 1993, 92(2), S. 308–316. Related Articles, Links
- P. S. Leung u. a.: Is there a relation between Chlamydia infection and primary biliary cirrhosis? In: Clin Dev Immunol., Juni–Dezember 2003, 10(2–4), S. 227–233.
- H. Y. Liu u. a.: The relationship between Chlamydia pneumoniae infection and primary biliary cirrhosis. In: Zhonghua Gan Zang Bing Za Zhi, September 2004, 12(9), S. 546–548.
- A. L. Mason u. a.: Pilot studies of single and combination antiretroviral therapy in patients with primary biliary cirrhosis. In: Am J Gastroenterol., Dezember 2004, 99(12), S. 2348–2355.
- H. T. Sørensen u. a.: Risk of primary biliary liver cirrhosis in patients with coeliac disease: Danish and Swedish cohort data. In: Gut, (Mai) 1999, 44, S. 736–738.
- L. Vilagut u. a.: Antibodies to mycobacterial 65-kD heat shock protein cross-react with the main mitochondrial antigens in patients with primary biliary cirrhosis. In: Eur J Clin Invest., August 1997, 27(8), S. 667–672. Related Articles, Links
- L. Xu u. a.: Cloning the human betaretrovirus proviral genome from patients with primary biliary cirrhosis. In: Hepatology, Januar 2004, 39(1), S. 151–156.
- T. Kumagi, E. J. Heathcote: Primary biliary cirrhosis. In: Orphanet J Rare Dis., 23. Januar 2008, 3, S. 1. PMID 18215315
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ Ulrich Beuers et al.: Changing nomenclature for PBC: From ‘cirrhosis’ to ‘cholangitis’. In: Clin Res Hepatol Gastroenterol. Band 39(5), Oktober 2015, S. e57–9, doi:10.1016/j.clinre.2015.08.001, PMID 26433440 (englisch).
- ↑ a b c Christian P. Strassburg et al.: S2k Leitlinie Autoimmune Lebererkrankungen. AWMF-Reg. Nr. 021-27. In: Z Gastroenterol. Band 55. Georg Thieme Verlag, 2017, S. 1135–1226, 1159–1160 (dgvs.de [PDF; abgerufen am 15. Januar 2021]).
- ↑ a b c d e f EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. In: Journal of Hepatology. 67j145–172, 2017 (englisch, easl.eu [PDF; abgerufen am 15. Januar 2021] European Association for the Study of the Liver – EASL). “The factors leading up to disease initiation are not well under-stood.”
- ↑ A. Lleo, C. Selmi, P. Invernizzi u. a.: Apotopes and the biliary specificity of primary biliary cirrhosis. In: Hepatology. Band 49, Nr. 3, März 2009, S. 871–879, doi:10.1002/hep.22736, PMID 19185000.
- ↑ K. Amano, P. S. Leung, R. Rieger u. a.: Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. In: J. Immunol. Band 174, Nr. 9, Mai 2005, S. 5874–5883, PMID 15845458 (jimmunol.org).
- ↑ J. S. English, R. J. Rycroft: Allergic contact dermatitis from methyl heptine and methyl octine carbonates. In: Contact Dermatitis. Band 18, Nr. 3, März 1988, S. 174–175, PMID 2966714.
- ↑ Kennzeichnung von Duftstoffen. In: bund.de. BVL, abgerufen am 13. Februar 2017.
- ↑ Klinische Studie (Phase 4): Phase 4 Study of Obeticholic Acid Evaluating Clinical Outcomes in Patients With Primary Biliary Cholangitis (COBALT) bei Clinicaltrials.gov der NIH
- ↑ EMA recommends revoking conditional marketing authorisation for Ocaliva. Europäische Arzneimittel-Agentur (EMA), 28. Juni 2024, abgerufen am 1. August 2024.
- ↑ Rote-Hand-Brief zu Ocaliva (Obeticholsäure): Widerruf der Genehmigung für das Inverkehrbringen in der Europäischen Union aufgrund eines nicht bestätigten klinischen Nutzens. Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), 1. August 2024, abgerufen am 1. August 2024.
- ↑ Übersicht von PBC-Studien. In: Clinicaltrials.gov. Abgerufen am 15. Januar 2021 (englisch).