Elektrophile aromatische Substitution
Eine elektrophile aromatische Substitution – abgekürzt als SEAr – ist eine elektrophile Substitutionsreaktion an einer aromatischen Verbindung. Während bei Aliphaten Substitutionen häufig nukleophiler Art sind, werden Aromaten bedingt durch ihr elektronenreiches π-System bevorzugt von Elektrophilen angegriffen. Dabei wird in der Regel ein an den reagierenden Aromaten gebundenes Wasserstoffatom durch das Elektrophil ersetzt. Die elektrophile aromatische Substitution ist eine mehrstufige Reaktion.
Reaktionsmechanismus[Bearbeiten | Quelltext bearbeiten]


Der Reaktionsmechanismus in Abbildung 1 beschreibt den allgemeinen Ablauf einer elektrophilen aromatischen Substitution. Der Aromat 1 steht in Wechselwirkung mit dem Elektrophil E+. Man spricht von der Bildung des π-Komplexes 2a. Aus diesem oder direkt aus den Ausgangsstoffen bildet sich unter Aufhebung der Aromatizität von 1 der mesomeriestabilisierte σ-Komplex 2b, der auch Arenium-Ion oder Wheland-Komplex genannt wird. Unter Deprotonierung dieses Komplexes findet eine Rearomatisierung des Systems statt, und das Endprodukt 3 wird freigesetzt. Die Bildung eines π-Komplexes ist nicht zwingend für die Erklärung der Reaktion notwendig.
Elektrophil[Bearbeiten | Quelltext bearbeiten]
Als Elektrophil E+ kommt ein weites Spektrum von Verbindungen in Frage, die häufig reaktive Produkte einer der elektrophilen aromatischen Substitution vorgelagerten Reaktion sind. Im Kontext der elektrophilen Substitution sind folgende Elektrophile von Bedeutung:
- das Proton
- polarisierte Halogene, z. B. Bromierung oder Iodierung:
- Br2 → Br+ + Br− unter Einwirkung einer Lewissäure als Katalysator
- Hypobromige Säure HBrO (Struktur H-O-Br)
- Iodchlorid I-Cl
- Carbokationen
- Carbonylverbindungen (C=O → Cδ+=Oδ−) unter Einwirkung einer Lewissäure als Katalysator bildet sich ein Acylkation
- Schwefeltrioxid in Schwefelsäure bildet HSO3+
- das Nitrosylkation
- das Nitryl- bzw. Nitronium-Kation (Nitrierung) aus [NO2]+[BF4]− oder durch Protolyse von Salpetersäure
Einige der Elektrophile sind positiv geladen und können direkt die Austrittsgruppe ersetzen. Bei anderen muss die positive Ladung durch Bindungsspaltung erzeugt werden. Die Koordination einer Lewissäure an das negative Ende des Elektrophils verstärkt die Polarisierung der Bindung und beschleunigt die elektrophile Substitution. Dies geschieht z. B. bei der Bromierung mit Brom, Br2, in Gegenwart von Eisen(III)-bromid, FeBr3.
Die Austrittsgruppe[Bearbeiten | Quelltext bearbeiten]
Im Regelfall ist die Austrittsgruppe ein Proton. Weiterhin sind auch Beispiele für Carbokationen, Sulfonyle und Silylgruppen bekannt.
Kinetik der Reaktion[Bearbeiten | Quelltext bearbeiten]
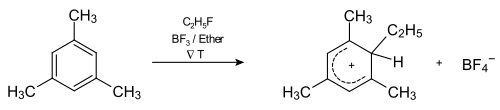
Abb. 3: Reaktionsgleichung zur Darstellung eines isolierbaren Wheland-Komplexes.

Abb. 2 zeigt die Reaktionskoordinate der Reaktion. Für die Zweistufigkeit gibt es viele Belege. Einerseits ist es möglich, die als Zwischenprodukt auftretenden Arenium-Ionen 2b in Reinsubstanz zu isolieren und zu charakterisieren. So gelingt die Darstellung des σ-Komplex 4 aus Mesitylen und Ethylfluorid in Gegenwart von Bortrifluorid in Ether bei −80 °C. Andererseits kann man über die Untersuchung von Isotopeneffekten eine Kinetik der Reaktion ermitteln, die für einen Zweistufenmechanismus spricht. Danach ist die Bildung des σ-Komplex der geschwindigkeitsbestimmende Schritt.
Substituenteneffekte[Bearbeiten | Quelltext bearbeiten]
Bereits am Aromaten vorhandene Substituenten üben einen großen Einfluss sowohl auf die Reaktivität des Aromaten und somit auf die Geschwindigkeit der elektrophilen Substitution, als auch auf die Position einer Zweitsubstitution aus (dirigierende Effekte). Wird die Elektronendichte des aromatischen Ringes erhöht, so nimmt auch seine Reaktivität gegenüber Elektrophilen zu. Diese Erhöhung kann durch mesomere und/oder induktive Effekte bewirkt werden – man spricht vom M- bzw. I-Effekt. Ein beigefügtes Vorzeichen zeigt an, ob der Substituent die Elektronendichte des Ringes erhöht (+M/+I-Effekt) oder verringert (−M/−I-Effekt).
Der induktive Effekt beruht darauf, dass elektronenziehende Substituenten den protonierten Ring destabilisieren. Der mesomere Effekt beruht darauf, dass der Substituent freie Elektronenpaare besitzt, über die er die Elektronendichte durch Mesomerie im Ring erhöhen oder vermindern kann.
Dirigierende Wirkung[Bearbeiten | Quelltext bearbeiten]
Neben der Beeinflussung der Reaktivität des Aromaten wirkt der Erstsubstituent dirigierend auf die Eintrittsposition des Zweitsubstituenten: Elektrophile aromatische Substitutionsreaktionen mit +I/+M-Substituenten liefern bezüglich dessen Stellung überwiegend ortho- und para-substituierte, −I/−M-Substituenten hingegen meta-substituierte Produkte.
Für eine Begründung dieses Effekts betrachtet man die mesomeren Grenzstrukturen entweder des aromatischen Reaktanten oder des σ-Komplexes. Tatsächlich entscheidend ist für die bevorzugte Zweitsubstitution die Höhe des ersten Übergangszustandes zwischen Edukt und dem σ-Komplex. Bei Betrachtung der mesomeren Grenzstrukturen des Eduktes argumentiert man mit einem erleichterten Angriff des Elektrophils an elektronenreichen Atomen des Rings und einem dadurch bedingten niedrigeren Übergangszustand. Bei der Betrachtung am σ-Komplex geht man davon aus, dass ein stabilerer, d. h. energieärmerer σ-Komplex auch über einen niedrigeren Übergangszustand erreicht wurde. Allgemein formuliert kann man sagen, dass elektronenspendende Gruppen – also Elektronendonatorgruppen – wie –CH3 oder –OCH3 ortho/para-dirigierend wirken, während elektronenanziehende Gruppen – sogenannte Elektronenakzeptoren – wie –NO2 oder –CO2H meta-dirigierend fungieren.
| Erstsubstituent | M-Effekt | I-Effekt | Dirigierender Effekt | Aktivierender Effekt |
|---|---|---|---|---|
| –O− | + | + | ortho / para | aktivierend stark |
| –OH / –NH2 / –NR2 | + | − | ortho / para | aktivierend stark |
| –OCH3 / –OR / –NHCOR | + | − | ortho / para | aktivierend mittel |
| Alkylrest | n/a | + | ortho / para | aktivierend schwach |
| –F / –Cl / –Br / –I | + | − | ortho / para | desaktivierend schwach |
| –CN / –COOH / –COOR / –COH / –COR | − | − | meta | desaktivierend mittel |
| –NO2 / –NR3+ / –CF3 / –CCl3 | − (nur –NO2) | − | meta | desaktivierend stark |
Zweitsubstitution[Bearbeiten | Quelltext bearbeiten]
Folgende Beispiele zeigen verschiedene Möglichkeiten der Zweitsubstitution:
Nitrotoluol

Nitrierung von Toluol

Bei der Nitrierung von Toluol mit Salpetersäure ist der +I-Effekt der Methylgruppe ausschlaggebend für die Steuerung des Zweitsubstituenten, so dass als Hauptprodukte o-Nitrotoluol mit 65 % und p-Nitrotoluol mit 30 % entstehen, m-Nitrotoluol dagegen nur zu 5 %.[1]
Dinitrobenzol

Nitrierung von Nitrobenzol

Hier bewirken der −I-Effekt und der −M-Effekt der Nitrogruppe des Nitrobenzols zu 93 % eine Steuerung in die meta-Stellung. Die ortho- und para-Stellungen treten nur zu 6 bzw. 1 % auf.[2]
Polysubstitution[Bearbeiten | Quelltext bearbeiten]
Bei einer elektrophilen aromatischen Substitution mit zwei oder mehr bereits vorhandenen Substituenten lässt sich die Reaktivität und der Ort der Substitution meist aus den kombinierten Effekten der Substituenten herleiten. Oft ist dies sehr einfach, wenn sich die Effekte der Substituenten miteinander verstärken oder alle freien Positionen äquivalent sind. Wenn dirigierende Wirkungen verschiedener Substituenten zu einer Substitution an verschiedenen Stellen führen würden, bestimmt normalerweise der am stärksten aktivierend wirkende Substituent, wo die Substitution stattfindet. Wenn zwei Positionen durch +I-Effekte von Alkylsubstituenten ähnlich bevorzugt werden, gewinnen sterische Effekte an Bedeutung und die Substitution findet an der leichter zugänglichen Position statt.[3]

Die Abbildung zeigt die Nitrierung von Nitrotoluol zu Dinitrotoluol. Zuerst bildet sich ein π-Komplex zwischen dem Nitroniumion NO2+ und dem Aromaten, dann reagieren diese zu einem auch als σ-Komplex bezeichneten Carbeniumion, dessen mesomere Grenzstrukturen in eckigen Klammern gezeigt werden. Dieses Zwischenprodukt ist im unteren Reaktionsweg weniger stabil als im unsubstituierten Aromaten, da die positive Ladung nahe der elektronenziehenden Nitro-Gruppe ist. Das obige Zwischenprodukt ist stabilisiert, da die positive Ladung nahe der elektronenschiebenden Methylgruppe ist. Das obere Produkt wird bevorzugt gebildet.
Elektrophile Substitutionsreaktionen an Aromaten (Auswahl)[Bearbeiten | Quelltext bearbeiten]
- Aminomethylierung
- Azokupplung
- Blanc-Reaktion (Chlormethylierung)
- Friedel-Crafts-Acylierung
- Friedel-Crafts-Alkylierung
- Halogenierung
- Houben-Hoesch-Synthese
- Hydroxymethylierung
- Gattermann-Synthese
- Gattermann-Koch-Synthese
- Kolbe-Schmitt-Reaktion
- Nitrierung
- Sulfonierung
- Vilsmeier-Haack-Reaktion
- Diazotierung
Einzelnachweise[Bearbeiten | Quelltext bearbeiten]
- ↑ Hans Beyer und Wolfgang Walter: Lehrbuch der Organischen Chemie, 19. Auflage, S. Hirzel Verlag, Stuttgart 1981, ISBN 3-7776-0356-2, S. 456.
- ↑ Joachim Buddrus: Grundlagen der organischen Chemie, 3. Auflage, Walter de Gruyter, 2003, ISBN 3-11-014683-5, S. 360.
- ↑ F. A. Carey: Organic Chemistry. 4. Auflage, McGraw-Hill 2000, ISBN 0-07-117499-0.