Lichtmikroskop

Lichtmikroskope (von griechisch μικρόν micrón „klein“, und σκοπεῖν skopein„etwas ansehen“) sind Mikroskope, die stark vergrößerte Bilder von kleinen Strukturen oder Objekten mit Hilfe von Licht erzeugen. Die Vergrößerung erfolgt gemäß den Gesetzen der Optik unter Ausnutzung von Lichtbrechung an Glaslinsen.
Um im erzeugten Bild Strukturen erkennen zu können, muss das Bild ausreichend Kontrast enthalten, der in vielen biologischen Objekten wie z. B. Gewebeschnitten oder kleinen Wasserlebewesen kaum vorhanden ist. Das ‚typische‘ mikroskopische Verfahren für solche Objekte ist die Hellfeldmikroskopie, bei der Kontrast durch farbige oder dunkle Strukturen im durchleuchteten Präparat hervorgerufen wird, bei Bedarf verstärkt durch zusätzliche künstliche Färbung des Objektes. Bei farblosen Präparaten kann Kontrast auch mit speziellen Beleuchtungsverfahren hervorgerufen werden, indem Unterschiede in der optischen Dichte (Brechungsindex) in Helligkeitsunterschiede umgewandelt werden. Dies geschieht bei Dunkelfeldmikroskopie, Phasenkontrastmikroskopie und bei Differentialinterferenzkontrast (DIC) oder bei dem bereits in den Anfängen der Mikroskopie verwendeten Verfahren mit schiefer Beleuchtung. Unterschiede im Polarisationsverhalten des Präparats werden bei der Polarisationsmikroskopie genutzt. Fluoreszente Strukturen im Präparat sind Voraussetzung für die Fluoreszenzmikroskopie und ihre zahlreichen Spezialverfahren. Weitere mikroskopische Verfahren sind die Konfokalmikroskopie und die Multiphotonenmikroskopie. All diese Verfahren sind in ihren eigenen Artikeln behandelt. Der Artikel hier stellt gemeinsame Grundlagen verschiedener mikroskopischer Verfahren dar.
Funktionsweise von einfachen und zusammengesetzten Mikroskopen[Bearbeiten | Quelltext bearbeiten]
Lichtmikroskopie kann mit „einfachen“ oder mit „zusammengesetzten“ Mikroskopen durchgeführt werden. Heutige Mikroskope sind typischerweise „zusammengesetzte Mikroskope“.
Einfache Mikroskope[Bearbeiten | Quelltext bearbeiten]

Einfache Mikroskope besitzen nur ein einzelnes optisches System zur Vergrößerung und funktionieren wie eine Lupe (zum Prinzip der Vergrößerung siehe dort). Ursprünglich verwendete man dazu nur eine einzelne Glaslinse. Um die für Mikroskope typische starke Vergrößerung zu erreichen, benötigt man eine sehr kurze Brennweite. Wegen der damit verbundenen starken Krümmung der Linsenoberfläche muss die Linse einen kleinen Durchmesser im Millimeterbereich haben. Sie muss mit entsprechend geringem Abstand dicht vor das Auge gehalten werden, was anstrengend ist und zur geringen allgemeinen Verbreitung dieser Mikroskope führte. Im einfachsten Fall bestand ein einfaches Mikroskop nur aus einer Glaslinse und einer Halterung für diese.
Die bekanntesten dürften jene von Antoni van Leeuwenhoek gebauten Geräte sein, mit denen eine mehr als 200-fache Vergrößerung erreicht wird. Damit gelangen ihm Ende des 17. Jahrhunderts zahlreiche wissenschaftliche Entdeckungen. Der in der Abbildung dargestellte Nachbau eines solchen Mikroskops wird mit der Seite, die auf der Unterlage liegt, dicht vor das Auge gehalten. Rechts im Bild ist am Ende einer Raute eine Spitze zu sehen, auf die das Präparat montiert wurde und mit der Spitze mittels eines Schraubgewindes in Position gebracht wurde. Darunter ist die Glaslinse in die Metallplatte eingelassen.
Im Laufe der Zeit wurden zahlreiche Varianten einfacher Mikroskope entwickelt wie z. B. das „Flohglas“, das Zirkelmikroskop, einfache Screw-barrel-Mikroskope, klassische Präpariermikroskope und die so genannten botanischen Mikroskope.[1] Auf der Suche nach besserer Abbildungsqualität verwendete man im 19. Jahrhundert auch Edelsteinlinsen wegen ihres hohen Brechungsindex und damit geringerer sphärischer Aberration oder eine Kombination aus zwei oder drei Plankonvexlinsen (Doublet, Triplet), ebenfalls zur Verringerung von Abbildungsfehlern. Es gab auch einfache Mikroskope mit kombinierbaren Einzellinsen mit Schraubfassung zur Änderung der Vergrößerung.[2] Solche einfachen Mikroskope wurden noch bis Ende des 19. Jahrhunderts angeboten. Zum Beispiel stellte Zeiss nach Firmengründung in Jena ab den 1850er Jahren Doublets bis 125facher Vergrößerung und Triplets bis 300facher Vergrößerung her,[3] 1895 noch ein Doublet 70fach.[4]
Da ein einfaches Mikroskop mit hoher Vergrößerung dicht vor das Auge gehalten werden muss, ist eine Beleuchtung des Präparats meist nur von der Rückseite möglich. Es wird dabei also in der Regel mit Durchlicht-Beleuchtung gearbeitet. Es gab aber seit der Erfindung eines die Linse umschließenden, zum Objekt gerichteten Beleuchtungs-Hohlspiegels durch Johann Lieberkühn (1740) auch die Möglichkeit der Auflichtbeleuchtung.[1] Die geringer vergrößernden damaligen einfachen "Naturforschermikroskope" waren meist Auflichtmikroskope.
Zweistufige Vergrößerung beim zusammengesetzten Mikroskop[Bearbeiten | Quelltext bearbeiten]

Zusammengesetzte Mikroskope bestehen aus mindestens zwei hintereinander geschalteten optischen Systemen mit jeweils eigener Vergrößerung. Das vordere, das Objektiv, erzeugt ein vergrößertes reelles Bild, das Zwischenbild, welches vom Okular ein zweites Mal vergrößert wird. Das Okular funktioniert dabei wie eine Lupe und erzeugt ein virtuelles Abbild des Zwischenbildes. Die Gesamtvergrößerung des Mikroskops ist das Produkt aus Objektivvergrößerung und Okularvergrößerung. Bei einem 20x Objektiv und einem 10x Okular beträgt die Gesamtvergrößerung also 200x.
Die ersten zusammengesetzten Mikroskope bestanden aus nur zwei Einzellinsen, sehr bald wurde das Okular zur Vergrößerung des nutzbaren Bildfeldes und Verringerung der Abbildungsfehler aus zwei Linsen zusammengesetzt (z. B. Huygens-Okular). In modernen Mikroskopen bestehen Objektive und Okulare aus mehreren Linsen, um verschiedene optische Abbildungsfehler auszugleichen. Hier ist etwa die chromatische Aberration zu nennen, die erst im 19. Jahrhundert durch Einführung neuer Glassorten begrenzt werden konnte. Da sich die Abbildungsfehler von Objektiv und Okular multiplizieren, waren zusammengesetzte Mikroskope den einfachen Mikroskopen zunächst unterlegen. Die Objektive und Okulare sind in der Regel wechselbar, so dass die Vergrößerung der jeweiligen Aufgabenstellung angepasst wird.
Bauweisen von zusammengesetzten Mikroskopen[Bearbeiten | Quelltext bearbeiten]
Durchlicht- oder Auflichtmikroskopie[Bearbeiten | Quelltext bearbeiten]
-
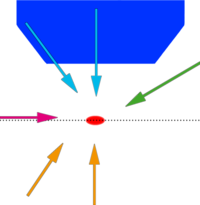 Beleuchtungsmöglichkeiten eines mikroskopischen Präparats (rotes Oval), das in der Schärfeebene (Punktlinie) vor dem Objektiv (dunkelblau) liegt (siehe Text).
Beleuchtungsmöglichkeiten eines mikroskopischen Präparats (rotes Oval), das in der Schärfeebene (Punktlinie) vor dem Objektiv (dunkelblau) liegt (siehe Text). -
 Robert Hooke arbeitete im 17. Jahrhundert mit einem Auflichtmikroskop.
Robert Hooke arbeitete im 17. Jahrhundert mit einem Auflichtmikroskop. -
 Insektenbeobachtung am Stereomikroskop mit seitlicher Auflicht-Beleuchtung.
Insektenbeobachtung am Stereomikroskop mit seitlicher Auflicht-Beleuchtung.


Je nachdem von welcher Seite das Licht auf das Präparat fällt, wird zwischen Auflicht- und Durchlicht-Beleuchtung beziehungsweise zwischen Auflicht- und Durchlichtmikroskopie unterschieden.
Bei der Durchlichtmikroskopie wird die Beleuchtung von hinten durch das Präparat hindurchgeleitet, bevor es vom Objektiv des Mikroskops aufgefangen wird (orange Pfeile in der Schemazeichnung). Daher sind durchsichtige oder dünn geschnittene Präparate erforderlich. Diese Technik wird beim häufigsten mikroskopischen Verfahren angewendet, der Durchlicht-Hellfeldmikroskopie.
Bei der Auflichtmikroskopie wird das Licht entweder vom Mikroskop kommend durch das Objektiv auf das Präparat geleitet (hellblaue Pfeile) oder von der Seite eingestrahlt (grüner Pfeil). Das vom Präparat reflektierte Licht wird wiederum vom Objektiv aufgefangen. Auflichtmikroskopie ist auch mit undurchsichtigen Präparaten möglich. Solche Präparate sind etwa in den Materialwissenschaften häufig, wo Probestücke eines Materials geschliffene und polierte oder auch angeätzte Oberflächen erhalten, die dann mikroskopisch untersucht werden. Auflichtbeleuchtung durch das Objektiv ist auch bei der Fluoreszenzmikroskopie weit verbreitet. Stereomikroskope arbeiten meist mit seitlicher Auflichtbeleuchtung.
Eine seitliche Beleuchtung (magenta-farbener Pfeil) wurde oder wird bei manchen Spezialverfahren verwendet (siehe Spaltultramikroskop und Lichtscheibenmikroskopie).
Aufbau eines typischen zusammengesetzten Durchlicht-Mikroskops[Bearbeiten | Quelltext bearbeiten]

Die Baugruppen eines typischen Durchlicht-Mikroskops wirken wie folgt zusammen:
- Das Objektiv (B) erzeugt ein reelles Bild, das Zwischenbild. Moderne Mikroskope sind meist mit mehreren Objektiven ausgestattet, die in einen Objektivrevolver montiert sind. Dies ermöglicht den schnellen Objektivwechsel durch Drehen des Revolvers.
- Das Zwischenbild wird vom Okular (A) ein weiteres Mal vergrößert. Die Zwischenbildebene liegt typischerweise innerhalb des Okulars. Die Gesamt-Vergrößerung des Mikroskops errechnet sich durch Multiplikation der Vergrößerungen von Objektiv und Okular. Viele Okulare haben eine 10-fache (10×) Vergrößerung. Häufige Objektiv-Vergrößerungen liegen zwischen 10× und 100×.
- Die Röhre zwischen Objektiv und Okular wird als Tubus bezeichnet.
- Das Präparat (auch: Objekt) ist bei Durchlicht-Mikroskopen üblicherweise auf dem gläsernen Objektträger (C) aufgebracht. Der Objektträger wird am Objekttisch (E) befestigt.
- Damit das von unten kommende Licht das Objekt optimal ausleuchtet, haben Durchlicht-Mikroskope ein gesondertes Linsensystem, den Kondensor (D). Dieser ist am Objekttisch befestigt.
- Der Objekttisch kann zum Scharfstellen des Objekts auf und ab bewegt werden. Der Kondensor wird dabei mitbewegt.
- Als Lichtquelle alter und sehr einfacher neuer Mikroskope dient ein Spiegel (F). Sonst wird eine elektrische Lichtquelle eingesetzt.
Die Beleuchtung des Präparats kann mittels kritischer Beleuchtung oder Köhlerscher Beleuchtung erfolgen (s. u.).
Tubuslänge, Endlichoptik und Unendlichoptik[Bearbeiten | Quelltext bearbeiten]

Die Objektive von älteren oder kleineren Mikroskopen sind angepasst an eine definierte Tubuslänge und erzeugen in einem genau definierten Abstand ein reelles Zwischenbild, das dann durch die Okularoptik vergrößert wird. Die Hersteller einigten sich auf eine Tubuslänge von 160 mm, bei älteren Mikroskopen kann diese Tubuslänge abweichen. So fertigte die Firma Leitz/Wetzlar nach einem hauseigenen Standard von 170 mm.
Diese definierte Tubuslänge bringt allerdings einige Nachteile mit sich. So können optische Elemente und Baugruppen nicht einfach in den Strahlgang eingefügt werden, da z. B. einfach der Platz hierfür nicht ausreichte. Neuere Mikroskope sind daher mit einer sogenannten „Unendlichoptik“ ausgestattet. In diesem Fall erzeugt das Objektiv kein reelles Zwischenbild, sondern das Licht verlässt das Objektiv als unendliche parallele Strahlen, was einen „unendlich“ langen Tubus ermöglicht. Somit können in den Strahlengang beliebig viele Zwischenelemente wie Filter, Strahlteiler etc. eingefügt werden. Da aus den parallel verlaufenden Lichtstrahlen kein Bild entstehen kann, befindet sich am Ende von unendlich-Tuben eine Tubuslinse. Diese erzeugt aus den parallelen Lichtstrahlen ein reelles Zwischenbild, das dann wieder durch die Okularoptik vergrößert werden kann. Unendlich Optik-Objektive erkennt man in der Regel an dem aufgebrachten ∞ (Unendlichzeichen).
In Abgrenzung zur Unendlichoptik wird die klassische Optik mit fester Tubuslänge als „Endlichoptik“ bezeichnet. Auf entsprechenden Objektiven ist die Länge des vorgesehenen Tubus in Millimetern angegeben, etwa 160 oder 170.
Aufrechte und inverse (auch: umgekehrte) Mikroskope[Bearbeiten | Quelltext bearbeiten]

Ein Mikroskop, bei dem sich das Objektiv oberhalb des Präparats befindet, wird als aufrechtes Mikroskop bezeichnet. Bei Durchlicht-Mikroskopen kommt das Licht dann von unten zum Präparat. Darüber ist das Objektiv, durch das das Licht nach oben zum Okular geht. Dies ist die häufigere Bauart.
Wird dieser Lichtweg umgekehrt, dann spricht man von einem inversen oder umgekehrten Mikroskop. Bei Durchlicht-Beleuchtung fällt das Licht hier von oben auf das Präparat, darunter befindet sich das Objektiv. Um ein bequemes Arbeiten zu ermöglichen, wird das Licht dann umgelenkt, so dass in die Okulare von oben hineingeschaut werden kann (siehe Abbildung).
Inverse Mikroskope werden beispielsweise zur Beobachtung von Zellkultur-Zellen eingesetzt, da sich die Zellen am Boden des Kulturgefäßes aufhalten. Der Abstand von den Zellen zum Objektiv wäre bei einem aufrechten Mikroskop zu groß. Mikroskope mit dieser Bauform sind ein unerlässliches Instrument für Untersuchungen an lebenden Zellen in Kulturgefäßen (Zellkultur), z. B. in der Patch-Clamp-Technik, sowie bei Einsatz von Mikromanipulatoren, die von oben an das Präparat herangeführt werden.
Beleuchtung des Präparats [Bearbeiten | Quelltext bearbeiten]
Um das Gesichtsfeld hell auszuleuchten, gibt es zwei verbreitete Beleuchtungsverfahren. Die kritische Beleuchtung ist die historisch ältere. Sie wird heute noch in manchen sehr einfachen Mikroskopen verwendet. Die von August Köhler entwickelte Köhlersche Beleuchtung erlaubt eine gleichmäßigere Beleuchtung des Präparats. Sie ist heute Standard in Routine- und Forschungsmikroskopen. Durchlicht-Hellfeldmikroskopie mit Köhlerscher Beleuchtung ist typischerweise der Ausgangspunkt für die Anwendung von speziellen lichtmikroskopischen Kontrastverfahren wie Phasenkontrast und Differentialinterferenzkontrast. Beide Beleuchtungsmethoden wurden ursprünglich für Durchlicht-Hellfeldmikroskopie entwickelt, werden aber auch in anderen Verfahren verwendet, wie der Fluoreszenzmikroskopie.
Kritische Beleuchtung[Bearbeiten | Quelltext bearbeiten]
Bei der kritischen Beleuchtung wird ein verkleinertes Abbild der Lichtquelle in der Präparateebene erzeugt. Wenn als Lichtquelle eine Glühlampe verwendet wird, wird also die Glühwendel mit Hilfe des Kondensors in der Schärfeebene abgebildet. Dadurch ist sichergestellt, dass das Präparat mit der maximal möglichen Helligkeit beleuchtet wird. Die Brennweite eines Mikroskopkondensors ist meist ziemlich kurz. Um ein Bild der Lichtquelle in der Schärfeebene des Mikroskops erzeugen zu können, muss erstens der Kondensor dicht am Präparat positioniert sein. Zweitens muss die Lichtquelle vergleichsweise weit vom Kondensor entfernt sein, so dass sie deutlich vor seiner vorderen Brennebene liegt. Um zu verhindern, dass das Bild der Glühwendel die Erkennung von Präparatstrukturen erschwert, wird unterhalb des Kondensors ein Mattglas-Filter in den Beleuchtungsstrahlengang gelegt. Sollte dies nicht ausreichend sein, kann der Kondensor etwas abgesenkt werden, so dass das Bild der Glühwendel unscharf wird.[5]

Wenn zur Beleuchtung keine Lampe, sondern ein Spiegel für Tageslicht eingesetzt wird, ist dieser meist auf einer Seite plan und auf der anderen Seite hohl. Der Hohlspiegel kann für Objektive mit geringer Vergrößerung eingesetzt werden, wenn der Kondensor entfernt wurde. Bei höheren Vergrößerungen muss die Beleuchtung auf einen kleineren Bereich des Präparats kondensiert werden. Dies geschieht mit dem Kondensor unter Verwendung des Planspiegels. Mit Tageslichtbeleuchtung kann es bei kritischer Beleuchtung zur Abbildung von Strukturen aus der Umgebung wie Fensterrahmen kommen. Auch hier hilft ein Mattglas-Filter unter dem Kondensor oder eine Kondensor-Absenkung.[5]
Köhlersche Beleuchtung [Bearbeiten | Quelltext bearbeiten]
August Köhler beschäftigte sich Ende des 19. Jahrhunderts mit der Mikrofotografie, also der Fotografie mit Hilfe eines Mikroskops. Bei direkter Beobachtung durch das Okular war die ungleichmäßige Helligkeit des Gesichtsfeldes bei kritischer Beleuchtung vergleichsweise wenig störend, da das Präparat je nach Bedarf hin und her verschoben werden konnte. Bei der Mikrofotografie führte eine ungleichmäßige Ausleuchtung jedoch zu einer schlechten Bildqualität. Er entwickelte daher ein Verfahren, das eine gleichmäßige Helligkeit bei gleich hoher Gesamthelligkeit erlaubte. Er veröffentlichte dieses Verfahren, das heute nach ihm benannt ist, 1893[7]. Der Vorgang des Einstellens der Köhlerschen Beleuchtung wird als köhlern bezeichnet.[5][6]
Köhlersche Beleuchtung hat neben einer gleichmäßigen Gesichtsfeldausleuchtung noch einen weiteren Vorteil: Nur der Bereich des Präparats, der tatsächlich beobachtet wird, wird beleuchtet. Dadurch wird störendes Streulicht, das in benachbarten Bereichen entstehen würde, vermieden. Bei dieser Beleuchtungsart wird ein Bild der Lichtquelle nicht in der Präparateebene erzeugt, sondern in der Ebene der Blende unterhalb des Kondensors. Diese wird als Kondensorblende oder Aperturblende bezeichnet. In der Präparateebene wird dagegen ein Bild der Leuchtfeldblende (auch: Feldblende) scharf abgebildet. Diese Blende befindet sich in der Nähe der Lichtquelle, in der Regel ist sie im Mikroskopfuß fest eingebaut. Das Bild in der Präparateebene wird scharfgestellt, indem der Kondensor auf oder ab bewegt wird. Köhlersche Beleuchtung ist nur mit einer künstlichen Lichtquelle möglich.[5][6]

Ein geköhlertes Mikroskop hat zwei miteinander in Beziehung stehende, verflochtene Strahlengänge und jeder der beiden hat mehrere 'konjugierte Ebenen', das heißt, was in einer der Ebenen scharf abgebildet ist, ist auch in den anderen konjugierten Ebenen scharf.
- Der Abbildungsstrahlengang (in der obigen Zeichnung der untere) hat folgende konjugierte Ebenen (in der Zeichnung bezeichnet mit Buchstaben im hellblau unterlegten Feld): Leuchtfeldblende (A), Präparatebene (B), Zwischenbild (C), Retina des Beobachters (D). Um dies zu erreichen, wird beim Vorgang des Köhlerns das Mikroskop zunächst auf zu beobachtende Strukturen im Präparat scharf gestellt, so dass diese im Zwischenbild und auf der Retina scharf sind. Dann wird die Leuchtfeldblende, die wie die Kondensorblende als Irisblende ausgeführt ist, zunächst zugezogen und der Kondensor wird in der Höhe so verstellt, dass der Leuchtfeldblendenrand ebenfalls scharf abgebildet wird. Falls nötig kann der Kondensor anschließend zentriert werden, so dass das Bild der Öffnung der Leuchtfeldblende genau in der Mitte des Gesichtsfeldes liegt. Anschließend wird die Leuchtfeldblende gerade so weit geöffnet, dass ihr Rand eben nicht mehr sichtbar ist.
- Der Beleuchtungsstrahlengang (in der Zeichnung oben) hat folgende konjugierte Ebenen (in der Zeichnung bezeichnet mit Ziffern im hellgrün unterlegten Feld): Lichtquelle (1), Kondensorblende (2), hintere Brennebene des Objektivs (3), Pupille des Beobachters (4).
Die Köhlersche Beleuchtung kann als eine kritische Beleuchtung angesehen werden, bei der die Lichtquelle die Öffnung der Leuchtfeldblende ist.[5][6]
Auflösung und Vergrößerung[Bearbeiten | Quelltext bearbeiten]


Bei optimaler Gerätebeschaffenheit und der Verwendung von Öl-Immersion lassen sich mit klassischer Lichtmikroskopie, wie sie im Wesentlichen im 19. Jahrhundert entwickelt wurde, bestenfalls Objekte voneinander unterscheiden, die 0,2 bis 0,3 µm oder weiter voneinander entfernt sind.[8] Die erzielbare Auflösung ist dabei nicht durch die verfügbare Qualität der Geräte, sondern durch physikalische Gesetze bestimmt. Sie hängt unter anderem von der Wellenlänge des verwendeten Lichts ab.
Verfahren, die seit den 1990er Jahren entwickelt wurden und auf nicht-linearen Farbstoffeigenschaften beruhen, erlauben auch eine Auflösung unter diesem so genannten Abbe-Limit.
Entscheidend für die Fähigkeit eines Mikroskops, Strukturen kleiner Objekte unterscheidbar abzubilden, ist (neben dem Kontrast) nicht die Vergrößerung, sondern die Auflösung. Dieser Zusammenhang ist nicht allein durch strahlenoptische Überlegung zu verstehen, sondern ergibt sich aus der Wellennatur des Lichts. Ernst Abbe erkannte als erster den entscheidenden Einfluss der Numerischen Apertur auf die Auflösung. Er gab als förderliche Vergrößerung

an. Dies bedeutet, dass die kleinsten vom Objektiv aufgelösten Strukturen nach der Abbildung durch das Okular im Auge noch aufgelöst werden können, also etwa unter einem Winkel von 2′ (Bogenminuten) erscheinen. Wird die Vergrößerung höher gewählt (z. B. durch ein Okular mit hoher Vergrößerung), wird das Bild des Objekts zwar noch größer dargestellt, aber es sind keine weiteren Objektdetails erkennbar. Objektive und Okulare müssen also aufeinander abgestimmt sein.
Nach den Gesetzen der Wellenoptik ist die Auflösung des Lichtmikroskops durch die Größe der Wellenlänge der Beleuchtung beschränkt, siehe Numerische Apertur.
Auflösungen jenseits des Abbe-Limits[Bearbeiten | Quelltext bearbeiten]

1971 veröffentlichten Thomas Cremer und Christoph Cremer theoretische Berechnungen über die Erzeugung eines idealen Hologramms zur Überwindung der Beugungsgrenze, das ein Interferenzfeld in allen Raumrichtungen festhält, ein sogenanntes -Hologramm.[9][10]

Seit den 1990er Jahren wurden einige Methoden entwickelt, die eine optische Auflösung jenseits des Abbe-Limits ermöglichen. Sie basieren alle auf Fluoreszenzmikroskopie und sind daher in diesem Artikel im Abschnitt Verfahren mit erhöhter Auflösung erwähnt.
Die folgenden neueren lichtmikroskopischen Entwicklungen erlauben eine Auflösung jenseits des klassischen Abbe-Limits:
- Stimulated Emission Depletion Microscope (STED)
- Photoactivated Localization Microscopy (PALM und STORM)
- 3D-SIM-Mikroskop
- 4Pi-Mikroskop
- TIRF-Mikroskop
- Vertico-SMI Strukturierte Beleuchtung SMI mit der SPDMphymod-Technologie (Lokalisationsmikroskopie-Basistechnologie)
- Optisches Rasternahfeldmikroskop (SNOM)
Verfahren zur Kontrastgewinnung[Bearbeiten | Quelltext bearbeiten]
- Hellfeldmikroskop, das „normale“ Lichtmikroskop
- Dunkelfeldmikroskop
- Phasenkontrastmikroskop
- Polarisationsmikroskop
- Differentialinterferenzkontrast
- Interferenzreflexionsmikroskop, auch Reflexionskontrast-Mikroskop genannt
- Kathodolumineszenzmikroskop
- Ultramikroskop
- Lichtscheiben-Mikroskopie (SPIM)
- Fluoreszenzmikroskop
- Konfokalmikroskop bzw. konfokales Laserscanningmikroskop (CLSM – Confocal Laser Scanning Microscope)
- Multiphotonenmikroskop einschließlich Zwei-Photonen-Mikroskop
Mikroskope für spezielle Anwendungen[Bearbeiten | Quelltext bearbeiten]
- Ein Stereomikroskop hat für beide Augen getrennte Strahlengänge, die das Präparat aus verschiedenen Winkeln zeigen, so dass ein dreidimensionaler Eindruck entsteht.
- Ein Strichmikroskop ist eine Ablesevorrichtung an einem Theodolit, einem Winkelmessgerät in der Vermessungskunde.
- Ein Operationsmikroskop wird von Ärzten im Operationssaal eingesetzt.
- Ein Trichinoskop wird bei der Fleischbeschau zum Nachweis von Trichinen (Fadenwürmer) eingesetzt.
- Ein Vibrationsmikroskop dient zur Untersuchung der Schwingung von Saiten bei Saiteninstrumenten.
- Ein Messmikroskop hat eine Zusatzeinrichtung, die eine Vermessung des Präparats erlaubt.
- Ein Computer-Mikroskop lässt sich zum Beispiel per USB-Kabel an einen Computer anschließen, der zur Anzeige der Abbildung benutzt wird.[11]
Geschichte[Bearbeiten | Quelltext bearbeiten]



Das Prinzip der Vergrößerung durch mit Wasser gefüllte Glasschalen wurde bereits von den Römern beschrieben (Seneca) und Vergrößerungslinsen waren schon im 16. Jahrhundert bekannt.
Der niederländische Brillenschleifer Hans Janssen und sein Sohn Zacharias Janssen werden oft als die Erfinder des ersten zusammengesetzten Mikroskops im Jahr 1590 angesehen. Dies basiert jedoch auf einer Erklärung von Zacharias Janssen selbst aus der Mitte des 17. Jahrhunderts. Das Datum ist dabei fragwürdig, da Zacharias Janssen selbst erst 1590 geboren wurde. Galileo Galilei entwickelte 1609 das Occhiolino, ein zusammengesetztes Mikroskop mit einer konvexen und einer konkaven Linse. Allerdings hatte Zacharias Janssen ein Gerät mit dem gleichen Funktionsprinzip bereits ein Jahr zuvor auf der Frankfurter Messe vorgeführt. Galileis Mikroskop wurde von der „Akademie der Luchse“ in Rom gefeiert, die im Jahr 1603 von Federico Cesi gegründet worden war. Eine Zeichnung des Akademiemitglieds Francesco Stelluti von 1630 gilt als älteste Zeichnung, die mit Hilfe eines Mikroskops angefertigt wurde. Auf ihr sind drei Ansichten von Bienen (von oben, unten und von der Seite) sowie Detailvergrößerungen zu sehen. Die Biene kam im Wappen der Familie Barberini vor, zu der Papst Urban VIII. gehörte. Stelluti schrieb in ein Banner oberhalb der Abbildung: „Für Urban VIII. Pontifex Optimus Maximus […] von der Akademie der Luchse, und in ewiger Verehrung widmen wir Euch dieses Symbol“.[12]
Ab etwa 1646 verwendete auch Athanasius Kircher ein Mikroskop, mit dem er Erreger der Pest zu erkennen glaubte.[13] Tatsächlich wurde Yersinia pestis jedoch erst Ende des 19. Jahrhunderts entdeckt.
Christiaan Huygens (1629–1695), ebenfalls Niederländer, entwickelte im späten 17. Jahrhundert ein einfaches Zwei-Linsen-Okularsystem. Es war bereits achromatisch korrigiert, hatte also weniger Farbfehler und war deshalb ein großer Fortschritt bei der Verbesserung der Optik im Mikroskop. Okulare nach Huygens werden bis heute produziert, sind jedoch im Vergleich zu modernen Weitfeldokularen optisch deutlich unterlegen.
Auch Robert Hooke benutzte für die Zeichnungen seiner 1665 publizierten Micrographia ein zusammengesetztes Mikroskop (siehe Abbildung). Die stärksten Vergrößerungen, die er in seinem Buch darstellte, waren 50-fach. Stärkere Vergrößerungen waren nicht möglich, da sich die Abbildungsfehler, die in der Frontlinse (Objektiv) und im Okular entstanden, vervielfachten, so dass keine feineren Details zu erkennen waren.

Antoni van Leeuwenhoek (1632–1723) verfolgte daher einen anderen Ansatz. Die Vergrößerung einer Linse ist umso stärker, je stärker sie gewölbt ist. Kleine, annähernd kugelförmige Linsen haben daher die stärkste Vergrößerung. Leeuwenhoek war brillant im exakten Schleifen kleinster Linsen, einer Technik, die zuvor nur unzureichend beherrscht worden war. Seine einfachen Mikroskope mit nur einer Linse waren zwar unhandlich zu benutzen, doch da er nur mit einer Linse mikroskopierte, entfiel die Multiplikation der Abbildungsfehler. Seine Mikroskope hatten eine bis zu 270-fache Vergrößerung.[14] So entdeckte Leeuwenhoek die von ihm so genannten „Animalkulen“, einzellige Bakterien und Protozoen.
Im Jahre 1768 beschrieb der Michel Ferdinand d’Albert d’Ailly, Duc de Chaulnes (1714–1769) das erste eigens für Messzwecke konzipierte Messmikroskop.
Robert Brown benutzte noch 1830 ein einfaches Mikroskop und entdeckte damit den Zellkern und die Brownsche Molekularbewegung. Es dauerte 160 Jahre, bevor zusammengesetzte Mikroskope dieselbe Abbildungsqualität erzeugten wie Leeuwenhoeks einfaches Mikroskop.
Bis weit ins 19. Jahrhundert hinein wurden gute zusammengesetzte Mikroskope durch Ausprobieren und anhand von Erfahrungswerten hergestellt. Ernst Abbe erarbeitete um 1873 die zum Bau besserer Mikroskope erforderlichen, noch heute gültigen physikalischen Grundlagen. Als Folge gelang es zum ersten Mal, ein Objektiv herzustellen, dessen Auflösungsgrenze nicht mehr durch die Materialgüte, sondern durch die physikalischen Beugungsgesetze limitiert wurde. Diese physikalische Auflösungsgrenze wird als das Abbe-Limit bezeichnet. Produziert wurden die entsprechenden Mikroskope zusammen mit Carl Zeiss in dessen optischen Werkstätten. Dabei profitierten sie von den von Otto Schott entwickelten optischen Gläsern und dem von August Köhler entwickelten Beleuchtungsapparat zur Köhlerschen Beleuchtung.
Siehe auch[Bearbeiten | Quelltext bearbeiten]
Literatur[Bearbeiten | Quelltext bearbeiten]
- Jörg Haus: Optische Mikroskopie. Wiley-VCH, Weinheim 2014, ISBN 978-3-527-41127-6. 220 Seiten.
- Michael Volger (Hrsg.: Irene K. Lichtscheidl): Lichtmikroskopie online. Abgerufen am 17. August 2018 (Theoretische Einführung und Anleitung zur praktischen Anwendung an der Uni Wien. Auch als pdf-Datei (270 Seiten) verfügbar.).
- Dieter Gerlach: Das Lichtmikroskop. Eine Einführung in Funktion, Handhabung und Spezialverfahren für Mediziner und Biologen. 2. Auflage. Georg Thieme Verlag, Stuttgart 1985, ISBN 3-13-530302-0.
Weblinks[Bearbeiten | Quelltext bearbeiten]
Zur Funktionsweise von Lichtmikroskopen:
- Theoretische Einführung und ausführliche Anleitungen zu einer Vielzahl mikroskopischer Techniken auf der Website der Uni Wien: Irene K. Lichtscheidl, Michael Volgger, et al.: Lichtmikroskopie online - Theorie und Anwendung. Core Facility Cell Imaging und Ultrastrukturforschung - Universität Wien, archiviert vom am 26. März 2023; abgerufen am 28. Oktober 2023.
- Verschiedene Kurzlehrgänge zur Lichtmikroskopie
- Optical Microscopy Primer: Umfangreiches Tutorial mit virtuellen Mikroskopen (englisch)
Sammlungen historischer Lichtmikroskope:
- Museum optischer Instrumente: Historische Mikroskope: Entwicklung des wissenschaftlichen Mikroskopbaus in Deutschland mit Geschichten zu ihren Herstellern und Anwendern, illustriert mit über 3000 Fotos
- Mikroskop-Museum: Die Geschichte des Lichtmikroskops von Anfang an bis heute in Wort und Bild. In der Galerie werden weit über 100 Mikroskope verschiedener Hersteller vorgestellt.
Einzelnachweise[Bearbeiten | Quelltext bearbeiten]
- ↑ a b Gerald Turner: Mikroskope. Verlag Callwey, München 1981, ISBN 978-3-7667-0561-7, S. 25–36.
- ↑ Dieter Gerlach: Geschichte der Mikroskopie. Verlag Harri Deutsch, Frankfurt am Main 2009, ISBN 978-3-8171-1781-9, S. 64–110, 171–179.
- ↑ Hermann Schacht: Das Mikroskop und seine Anwendung. Verlag G. W. F. Müller, Berlin 1851, Kapitel: II. 2.
- ↑ Zeiss Jena: Katalog No. 30: Mikroskope und mikroskopische Hilfsapparate. Eigenverlag, Jena 1895, S. 105.
- ↑ a b c d e Dieter Gerlach: Das Lichtmikroskop. Eine Einführung in Funktion, Handhabung und Spezialverfahren für Mediziner und Biologen. Georg Thieme Verlag, Stuttgart 1976, ISBN 3-13-530301-2, S. 64–71.
- ↑ a b c d Jörg Haus: Optische Mikroskopie Funktionsweise und Kontrastierverfahren. John Wiley & Sons, 2014, ISBN 978-3-527-41286-0, S. 17–21 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ August Köhler: Ein neues Beleuchtungsverfahren für mikrophotographische Zwecke. In: Zeitschrift für wissenschaftliche Mikroskopie. Band X., Nr. 4, 1893, S. 433–440 (online bei archive.org).
- ↑ Ernst Abbe: Beiträge zur Theorie des Mikroskops und der Mikroskopischen Wahrnehmung. In: Archiv für Mikroskopische Anatomie. 9, 1873, S. 413–468.
- ↑ Patentanmeldung DE2116521A1: Verfahren zur Darstellung bzw. Modifikation von Objekt-Details, deren Abmessungen außerhalb der sichtbaren Wellenlängen liegen. Angemeldet am 5. April 1971, veröffentlicht am 12. Oktober 1972, Erfinder: Christop Cremer, Thomas Cremer.
- ↑ Konstruktionsplan 1978: Konfokales Laser Scanning Fluoreszenzmikroskop mit hoher Auflösung und Schärfentiefe/4Pi Point Hologram ( des vom 4. März 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. (PDF; 83 kB).
- ↑ Computermikroskop: Die bessere Webcam, test.de, 23. Januar 2003 (online abgerufen am 26. Februar 2013).
- ↑ Stephen Jay Gould: Die Lügensteine von Marrakesch: Vorletzte Erkundungen der Naturgeschichte. S. Fischer, Frankfurt 2003, ISBN 3-10-027813-5, S. 52–53.
- ↑ Luigi Belloni: Athanasius Kircher. Sein Mikroskop, die Animalcula und Pestwürmer. In: Medizinhistorisches Journal. Band 20, 1985, S. 58–65.
- ↑ Hugo Freund, Alexander Berg: Geschichte der Mikroskopie. Leben und Werk großer Forscher. Band I: Biologie. Umschau Verlag, Frankfurt am Main 1963, S. 4–5.