Guanidin
| Strukturformel | |||||||
|---|---|---|---|---|---|---|---|

| |||||||
| Allgemeines | |||||||
| Name | Guanidin | ||||||
| Andere Namen |
| ||||||
| Summenformel | CN3H5 | ||||||
| Kurzbeschreibung |
farblose, hygroskopische Kristalle [1] | ||||||
| Externe Identifikatoren/Datenbanken | |||||||
| |||||||
| Eigenschaften | |||||||
| Molare Masse | 59,07 g·mol−1 | ||||||
| Aggregatzustand |
fest | ||||||
| Schmelzpunkt | |||||||
| Löslichkeit |
leicht löslich in Ethanol, in Wasser Protonierung zu leichtlöslichem Guanidinium[1] | ||||||
| Sicherheitshinweise | |||||||
| |||||||
| Toxikologische Daten | |||||||
| Thermodynamische Eigenschaften | |||||||
| ΔHf0 |
−56,0 kJ/mol[4] | ||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||
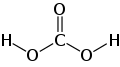
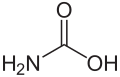
Guanidin ist eine chemische Verbindung an der Grenze zwischen anorganischer und organischer Chemie. Guanidin kann als Stickstoffanalogon der Kohlensäure aufgefasst werden, ähnlich wie Carbamidsäure und Harnstoff als entsprechende Zwischenstufen.[5]
Guanidin wurde schon in der Mitte des 19. Jahrhunderts zum ersten Mal synthetisiert,[6] die Kristallstruktur konnte aber erst 2009 gelöst werden.[7] Es wurde vermutet,[7] dass so viel Zeit vergehen musste, da Guanidin (im offensichtlichen Gegensatz zur Kohlensäure) eine der stärksten organischen Basen ist[8] und an Luft spontan mit Luftfeuchtigkeit und Kohlenstoffdioxid zu Guanidiniumcarbonat reagiert.[6]
Guanidin wurde in verschiedenen quantenchemischen Rechnungen untersucht, vor allem in Bezug auf das Konzept der Y-Aromatizität.[9] Des Weiteren stellt Guanidin eine wichtige Substruktur in vielen biologischen Molekülen wie Guanin, Arginin oder Guanosin dar.[7]
Geschichte
Guanidin wurde erstmals 1861 durch oxidativen Abbau von Guanin synthetisiert.[6] Röntgenographische Strukturdaten von Addukten des Guanidins wurden 2007 erhalten,[10] doch gelang die vollständige Aufklärung der Guanidin-Kristallstruktur trotz der einfachen Molekülstruktur erst 148 Jahre nach der ersten Synthese.[7] Schließlich wurde 2013 die Position der Wasserstoffatome mittels Neutronenbeugung am Guanidin-Einkristall sehr genau bestimmt.[11]
Vorkommen
Viele Naturstoffe sind Guanidinderivate, darunter so wichtige wie die proteinogene Aminosäure Arginin, das Kreatin und das Kreatinin. Die Guanidinderivate Arginin und Argininosuccinat spielen eine wichtige Rolle im Harnstoffzyklus und damit bei der Entgiftung des durch Stoffwechselprozesse gebildeten Ammoniaks.
Chemische Eigenschaften
Guanidin ist in Wasser eine extrem starke Base mit einem pKB-Wert von 0,30 und ist damit vergleichbar basisch wie ein Alkalihydroxid.[12] Vor der Entdeckung von Protonenschwämmen wurde Guanidin als die stärkste organische Base betrachtet.[13] Deshalb bildet sich sofort bei Kontakt mit Feuchtigkeit, z. B. mit Luftfeuchtigkeit, das Guanidinium-Kation [C(NH2)3]+. Die Reaktivität ist ausreichend, um Kohlenstoffdioxid aus der Luft zu binden im Guanidiniumcarbonat.[6] Die Basizität in der Gasphase wurde hingegen als deutlich kleiner vorhergesagt.[14]

Diese hohe Reaktivität von Guanidin und die sehr große Stabilität des Guanidinium-Kations wurde mit unterschiedlichen Konzepten zu erklären versucht. Ein einfacher Vorschlag war die größere Mesomeriestabilisierung des Guanidiniumions gegenüber der freien Base. Darauf baut das Konzept der Y-Aromatizität auf; das Guanidiniumion ist zwar nicht cyclisch, besitzt aber sechs π-Elektronen, die über das Molekül delokalisiert sind. Dies solle zu einer Stabilisierung führen. Andere Arbeiten sahen hingegen die vorherrschende Stabilisierung in den zahlreichen Wasserstoffbrückenbindungen zwischen Guanidinium und Wassermolekülen.[9][13][14]
Guanidin kann als strukturell verwandt mit der Kohlensäure begriffen werden, wobei die Hydroxygruppen durch Aminogruppen und die Carbonylgruppe durch eine Iminogruppe ersetzt wurden. Entsprechend sind Carbamidsäure und Harnstoff Zwischenstufen dieser Stickstoff- und Sauerstoffanaloge.[5]
-
-
-

-
 Guanidin
Guanidin



Gewinnung und Darstellung
Adolph Strecker synthetisierte Guanidin aus Guanidiniumsulfat, das er durch oxidativen Abbau aus Guanin gewann. Dazu versetzte er das Salz der Schwefelsäure mit Barytwasser (einer Bariumhydroxid-Lösung) und verdunstete das Lösungsmittel im Vakuum. Aufgrund der hohen Hygroskopie des Guanidins konnte er die freie Base jedoch nicht untersuchen.[6]
Eine andere Synthesevorschrift nutzte eine Metathese-Reaktion, um Guanidin zu erhalten. Dafür wurde Kaliumhydroxid mit Guanidiniumperchlorat stöchiometrisch in Ethanol umgesetzt. Das entstandene Guanidiniumhydroxid wurde über Phosphorpentoxid im Vakuum getrocknet, um Wasser abzuspalten. Ein IR-Spektrum des so synthetisierten Guanidin lieferte erste Belege für die Molekülstruktur.[15][16]
Um Guanidin zu kristallisieren, wurde Guanidiniumchlorid in THF gelöst und mit einer Natriummethanolatlösung ebenfalls in THF unter Luftausschluss versetzt. Zu der Lösung konnte langsam Acetonitril diffundieren. Dabei bildeten sich Einkristalle des Addukts von Guanidin und 3-Amino-5,6-dimethyl-[1,2,4]triazin, letzteres durch eine Reaktion des Acetonitrils mit Guanidin in der Gegenwart des Alkoholats. So konnte Guanidin als Molekül zum ersten Mal röntgenographisch untersucht werden.[10]
Mit einer ähnlichen Metathese-Reaktion konnte die reine, freie Base Guanidin synthetisiert werden. Dazu wurde Guanidiniumchlorid mit Natriumethanolat in Ethanol – unter Schutzgasatmosphäre – umgesetzt, wobei Natriumchlorid ausfiel (siehe Reaktionsgleichung). Die Lösung wurde gefiltert, das Lösungsmittel konnte langsam verdampfen. Beim Abkühlen fielen Einkristalle aus.[7]

Ein für die Neutronenbeugung ausreichend großer Einkristall wurde gezüchtet, indem eine Guanidin-Lösung in Ethanol etwa ein halbes Jahr auskristallisieren konnte.[11]
Derivate
- Guanidin bildet mit Säuren Guanidinium-Salze, z. B. Guanidiniumchlorid, Guanidiniumthiocyanat, Guanidiniumnitrat und Guanindiniumcarbonat
- Guanidin-Derivate werden zur Herstellung von Flammschutzmitteln und Harzen verwendet. Cocospropylendiaminguanidiniumacetat für Desinfektionsmittel.
- Vom Guanidin leitet sich eine Reihe von Sprengstoffen ab, so z. B.:
- Guanidiniumnitrat
- Nitroguanidin
- Aminonitroguanidin
- Amino-, Diamino-, Triaminoguanidin und deren Salze
- Dinitroguanidin und seine Salze
- Tetrazen
- Als Arzneimittel findet das Biguanidin Metformin bei der Therapie von Typ 2 Diabetes Verwendung
- Eine Reihe von Derivaten des Guanidins hat einen extrem süßen Geschmack, bis zur 200.000-fachen Süßkraft der Saccharose. Sie gehören damit zu den süßesten bisher bekannten Verbindungen.[17]
Einzelnachweise
- ↑ a b c Eintrag zu Guanidin. In: Römpp Online. Georg Thieme Verlag
- ↑ Dieser Stoff wurde in Bezug auf seine Gefährlichkeit entweder noch nicht eingestuft oder eine verlässliche und zitierfähige Quelle hierzu wurde noch nicht gefunden.
- ↑ Eintrag in der ChemIDplus-Datenbank der United States National Library of Medicine (NLM) (Seite nicht mehr abrufbar)
- ↑ David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Standard Thermodynamic Properties of Chemical Substances, S. 5-20.
- ↑ a b Edward C. Franklin: The Ammono Carbonic Acids. In: Journal of the American Chemical Society. 44. Jahrgang, 1922, S. 486–509, doi:10.1021/ja01424a007.
- ↑ a b c d e A. Strecker, Liebigs Ann. Chem. 1861, 118, 151.
- ↑ a b c d e T. Yamada, X. Liu, U. Englert, H. Yamane, R. Dronskowski: Solid-State Structure of Free Base Guaninide Achieved at Last. In: Chem. Eur. J. 15. Jahrgang, 2009, S. 5651, doi:10.1002/chem.200900508.
- ↑ S. J. Angyal, W. K. Warburton: The basic strengths of methylated guanidines. In: Journal of the Chemical Society (Resumed). 1951, S. 2492–2494, doi:10.1039/jr9510002492.
- ↑ a b R. Caminiti, A. Pieretti, L. Bencivenni, F. Ramondo, N. Sanna: Amidine N−C(N)−N Skeleton: Its Structure in Isolated and Hydrogen-Bonded Guanidines from ab Initio Calculations. In: The Journal of Physical Chemistry. 100. Jahrgang, 1996, S. 10928–10935, doi:10.1021/jp960311p.
- ↑ a b M. Goebel, T.M. Klapoetke: First structural characterization of guanidine. In: Chem. Commun. 43. Jahrgang, Nr. 30, 2007, S. 3180–3182, doi:10.1039/B705100J.
- ↑ a b P. K. Sawinski, M. Meven, U. Englert, R. Dronskowski: Single-Crystal Neutron Diffraction Study on Guanidine, CN3H5. In: Cryst Growth Des. 13. Jahrgang, 2013, S. 1730–1735, doi:10.1021/cg400054k.
- ↑ H. R. Christen, F. Vögtle: Organische Chemie - Von den Grundlagen zur Forschung. 2. Auflage, S. 425, Otto Salle Verlag, Frankfurt a. Main 1996, ISBN 3-7935-5398-1.
- ↑ a b Alberto Gobbi, Gernot Frenking: Y-Conjugated compounds: the equilibrium geometries and electronic structures of guanidine, guanidinium cation, urea, and 1,1-diaminoethylene. In: Journal of the American Chemical Society. 115. Jahrgang, 1993, S. 2362–2372, doi:10.1021/ja00059a035.
- ↑ a b Kenneth B. Wiberg: Resonance interactions in acyclic systems. 2. Y-Conjugated anions and cations. In: Journal of the American Chemical Society. 112. Jahrgang, 1990, S. 4177–4182, doi:10.1021/ja00167a011.
- ↑ W. Marckwald, F. Struwe: Über einige Guanidoniumsalze. In: Ber. 55. Jahrgang, 1922, S. 457–463, doi:10.1002/cber.19220550221.
- ↑ W. Jeremy Jones: The infra-red spectrum and structure of guanidine. In: Trans. Faraday Soc. 55. Jahrgang, 1959, S. 524–531, doi:10.1039/TF9595500524.
- ↑ H.-D. Belitz et al.: Lehrbuch der Lebensmittelchemie. 5. Aufl., Springer, Berlin u. a. 2001. S. 433.